|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
�t�H���E�q�b�y���E�����h�E(von Hippel-Lindau�GVHL)�a(���邢�͏nj�Q)(MIM ID#193300)�́A����F�̗D����`���̎����ŁA�����̑���Ɏ�ᇐ����邢�͔X�E���a�ς𑽔�����B���Ǖa�ςƂ��ẮA�Ԗ����ǎ�A�����_�o�n(���]�A�����A�Ґ�)�̌��lj��A�X���̐_�o�������ᇁE�X�E�A���t���F�זE��A�t���̎�ᇁE�X�E�A������̔X�E�B��A����ɓ��������p�X�̎�ᇂ⏗���̎q�{�L�Ԗ��̔X�E�B��Ȃǂ�����Ă���B
���j�I�ɂ́A�h�C�c�̊�Ȉ�ł���Eugen von Hippel���Ԗ��̑������ǎ��A�Ƒ���ɒ��ڂ��A19���I������20���I�����ɂ����Ă�������Ă���1, 2)�B�܂��X�E�F�[�f���̐_�o�a����ł���Arvid Lindau�́A�Ԗ��݂̂łȂ������_�o�n�ɂ����ǎ�𑽔�����Ƒ���̕a���������������3, 4)�B���̌�{�����̗Տ��a�Ԃ��AMelmon��A�����Lamiell��ɂ���Đ�������A�{�����͐��2�l�̈�t����������von Hippel-Lindau�a�Ƃ���悤�ɂȂ��Ă���5, 6)�B1988�N��Seizinger��͉ƌn�̘A����͂ɂ��A�q�g���F��3�ԒZ�r��Ɍ�����`�q�̋Ǎ݂𐄒肵��7)�B����5�N��ɁA�č�NIH/NCI�̃O���[�v�����S�ƂȂ�Apositional cloning�@�ɂ��3p25-26�̈��茴����`�q�̓���ɐ������Avon Hippel-Lindau�a(VHL)��`�q�Ƃ���1993�N�ɕ���8)�B
 |
 |
| Eugen von Hippel |
Arvid Lindau |
- von Hippel E. Vorstellung eines Patienten mit einer sehr ungewohnlichen Netzhaut. Ber Deutsch Ophthal Ges. 1895�G24�F269.
- von Hippel E. Uber eine sehr seltene Erkrankung der Netzhaut. Albrecht von Graefes Arch Ophthal. 1904�G59�F83-106.
- Lindau A. Studien uiber Kleinhirncysten. Bau, Pathogeneseund. Beziehungen fur angiomatosis retinae. Acta Pathol Microbiol Scand. 1926�F3(Suppl 1)�G1-128.
- Lindau A. Zur Frage der Angiomatosis Retinae und Ihrer Hirncomplikation. Acta Ophthal. 1927�G4�F193-226.
- Melmon KL, Rosen SW. Lindau's disease: review of the literature and study of a large kindred. Am J Med. 1964�G36�F595-617.
- Lamiell JM, Salazar FG, Hsia YE. Von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine(Baltimore). 1989�G68(1)�F1-29.
- Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988�G332(6161)�F268-9.
- Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993�G260(5112)�F1317-20.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
VHL��`�q�͊��}����`�q(tumor suppressor gene)�ɕ��ނ���AKnudson������2-hit�̋@�\��2�̃A����(allele)�ɕψق��N���邱�Ƃł��̋@�\���������A�זE�̎�ᇉ����n�܂�ƍl������BVHL�ƌn���҂ł́A��`�I�ψ�(germline mutation)�ɂ��A�o�����ɂ��łɕБ���VHL��`�q�̕s���������N�����Ă���(1-hit)�A���̌�Η�allele�ɑ̍זE�ψ�(somatic mutation)���N���邱�Ƃ�(2-hit)�A��`�q�@�\�����S�ɏ�������B����A�U����̐t��ᇂȂǂł�VHL��`�q�̍��p�x�̕ψفA�s�����������o����邪�A���̏ꍇ�ɂ́A2��̑̍זE�ψق��N���Ă���B�Տ��I��VHL�a�Ɛf�f���ꂽ�ƌn���҂ɂ����Ă�80�`90%�ŁA���̈�`�q�̈�`�I�ψق����o�ł���̂ŁA���̈�`�q�ψق��w�W�ɂ����A�������`�q�f�f(DNA test)���s���Ă���B
VHL��`�q��3��exon���\������Ă���A�q�g�Q�m����ł�3p25.3��̖�13,000bp�̗̈�ɑ��݂��A��������S����4.5kb��mRNA���]�ʂ����1)�BmRNA�̒`���|��̈��639����ł��邪�A�A�~�m�_1�Ԃ�54�Ԃ�2�J���̃��`�I�j�����|�J�n����A���ꂼ��213��160�A�~�m�_(��30kd��19kd�̃T�C�Y)��VHL�`��������A���҂Ƃ���ᇗ}���@�\�������Ă���2, 3)�B
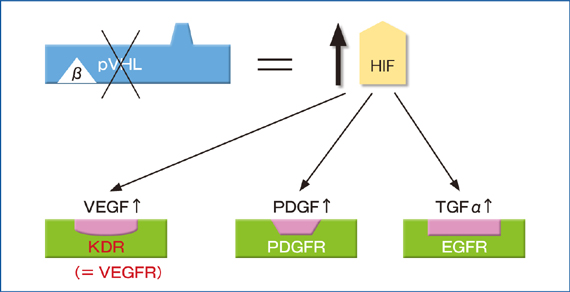
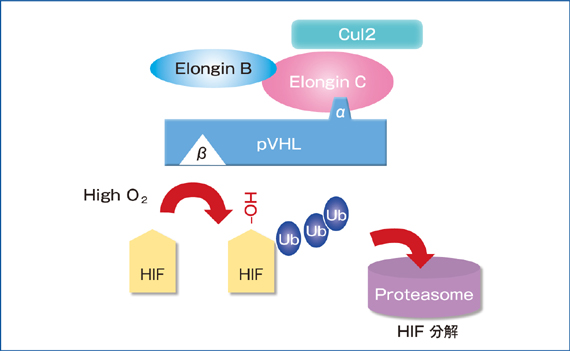
VHL�`��(pVHL)�̋@�\�ł���܂łɍł��悭��͂���Ă���̂��AE3 ubiquitin ligase�����̂Ƃ��Ă̋@�\�ł���A�]�ʈ��qHIF(hypoxia-inducible factor)(��_�f�U�����q)�̕��𐧌���s���Ă���BpVHL�̓��A����2�̍\���@�\�̈�(domain)����Ȃ�A��-domain��Elongin C�A�����Elongin B�ACUL2�ARBX1�ƌ������AE3 ubiqutin ligase������(VHL/E3 complex)���`������4-6)�B��������̃�-domain�ŕW�I�`���ƌ������邪�A���̃��r�L�`�����W�I�`����1���A�|���C��(�v�������c��̐��_��)����HIF���ł���B�]�ʈ��qHIF��HIF����HIF����2���q�̃w�e�������̂��`�����A�����HIF����cofactor�ł���CBP/p300���������A�]�ʈ��q�Ƃ��ċ@�\���������BHIF���͐���_�f����Ԃł�HIF prolyl hydroxylase(HPH)�ɂ��v�������c��(HIF1���ł�402�A564�ԁAHIF2���ł�405�A531�Ԃ̃A�~�m�_)�����_������|���C������BHPH�ɂ�萅�_��(�|���C��)���ꂽHIF���`����VHL/E3 complex�Ń|�����r�L�`��������A���̌�26S proteasome�ŋ}���ɕ��������7, 8)�B����A��_�f��Ԃł�HIF���̃��r�L�`�����ƕ������}������AHIF���͊j���Ɉڍs��HIF���ƌ������A��`�qpromoter����HRE(hypoxia response element)�Ɍ������l�X�Ȉ�`�q�̓]�ʂ𑣐i����9)�B
HIF�ɂ��]�ʂ�����`�q�͂���܂ł�100�ȏオ�m���Ă���A�@���ǐV���A�A�זE���A�V�h�[�V�X��A�B�O���R�[�X�̎�荞�݁E���C�I�n�̑��i�A�N�G���_��H�̗}���A�C�זE�ڒ����̒ቺ�A�^�����E�]�ڔ\�̑��i�A�}�g���b�N�X�̍č\���A�ȂǗl�X�ȋ@�\�ɂ�������Ă���9-12)�B����AVHL���s�����������זE�ł́A����_�f����Ԃɂ����Ă�HIF���̕������ł����AHIF�͂����̈�`�q�Q���P��I�A���I�ɔ��������A���ꂪ�זE�̎�ᇉ��Ɍ��т��Ă��邱�Ƃ��z�肳��Ă���B�@�Ɋ֘A�����`�q�Ƃ��ẮAVEGF�APDGFB�AANGPT2�Ȃǂ��m���Ă���A���ǂ̓���זE�����זE(pericyte)�̑��B�𑣐i���A���ǂ̐V���E���n�E�ێ��Ȃǂ̍�p�����BVHL�a�œ����I�Ȍ��lj���t��ᇂł͎�ᇌ��ǂ̑����������ł���AVEGF�����������Ă���B
����ɁAVHL�`����HIF���߈ȊO�ɂ��l�X�ȋ@�\�������Ƃ��z�肳��Ă���Ai) �_�o�זE��apoptosis�}���Ɗ��F�זE��̔����@�\�Aii) fibronectin(FN1)�Atype IV collagen�Ƃ̌����ƍזE�O�}�g���b�N�X�̍\�����߁Aiii) �זE��primary cilia�̌`���ƔX�E�`���A�Ȃǂɂ��Ă����݉�͂��i�݂���13, 14)�B
 |
| �}2-1 VHL�a�ɂ������ᇔ��ǂ̋@�\ |
 |
| �}2-2 VHL�`���̕����̂ɂ��HIF�̕��� |
- Renbaum P, Duh FM, Latif F, et al. Isolation and characterization of the full-length 3' untranslated region of the human von Hippel-Lindau tumor suppressor gene. Hum Genet. 1996�G98(6)�F666-71.
- Iliopoulos O, Kibel A, Gray S, et al. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med. 1995�G1(8)�F822-6.
- Schoenfeld A, Davidowitz EJ, Burk RD. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc Natl Acad Sci USA. 1998�G95(15)�F8817-22.
- Kamura T, Koepp DM, Conrad MN, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999�G284(5414)�F657-61.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia inducible factors for oxygen-dependent proteolysis. Nature. 1999�G399(6733)�F271-5.
- Stebbins CE, Kaelin WG Jr, Pavletich NP. Structure of the VHL-Elongin C-Elongin B complex: implications for VHL tumor suppressor function. Science. 1999�G284(5413)�F455-61.
- Ivan M, Kondo K, Yang H, et al. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001�G292(5516)�F464-8.
- Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2 regulated prolyl hydroxylation. Science. 2001�G292(5516)�F468-72.
- Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006�G441(7092)�F437-43.
- Kelly BD, Hackett SF, Hirota K, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003�G93(11)�F1074-81.
- Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004�G10(8)�F858-64.
- Manalo DJ, Rowan A, Lavoie T, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005�G105(2)�F659-69.
- Frew IJ, Krek W. pVHL�Fa multipurpose adaptor protein. Sci Signal. 2008�G1(24)�Fpe30.
- Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein�FO2 sensing and cancer. Nat Rev Cancer. 2008�G8(11)�F865-73.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
�����_�o�n(�]�Ґ�)���lj��A�Ԗ�����(��)��A���������p�X��A�X�X�E�A�X�_�o�������ᇁA�t�X�E�A�t��ᇁA���F�זE��A������̔X��A�q�{�L�Ԗ��X��Ȃǂ����ǂ���B�\1�ɊC�O�A��ɕč��ł̔��ǔN��Ɣ��Ǖp�x�������B���ǂ����ᇂ͂ǂ���������ōĔ����A��N���ǂƂ��������������Ă���B�T�^�͒����_�o�n���lj��ł��葽�����A�Ĕ����Ő_�o�Ǐ�������A���҂�QOL�̒������ቺ���N�����B�t��ᇂ��X�_�o�������ᇂ́A���������Ĕ����ł���B�{�M�ɂ�����e��ᇂƔX�E�̔��Ǖp�x�Ɗ��Ґ��͏ڍׂȒ������ʂ��Ȃ����ߕs���ł���B
| �\1 VHL�a�Ŕ��ǂ����� |
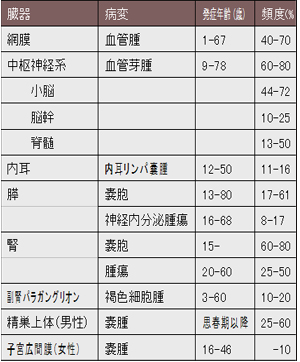 |
���F�{�M�ɂ�����e��ᇂƔX�E�̔��Ǖp�x�͒������ʂ��Ȃ����ߕs���ł���B
(Lonser R, et al. Lancet. 2003�G361�F2059-67)1)
|
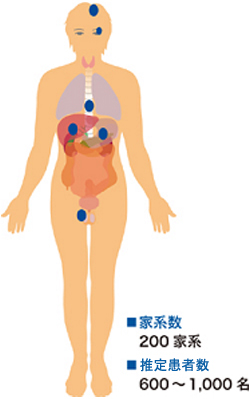 |
�}3 VHL�a�Ŏ�ᇂ����ǂ��鑟��ƕp�x
�@�@���F���Ǖ��� |
|
- Lonser R, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361�F2059-67.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
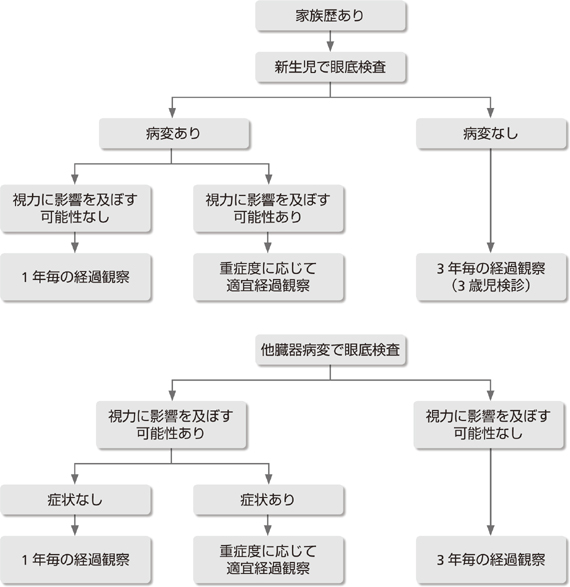
�@ VHL�a�̉Ƒ��������炩�ł���ꍇ
�Ԗ����ǎ�A�����_�o�n���lj��A���������p�X��A�t��ᇁA���F�זE��A�X���̕a�C(�X�X�E�E�X���̐_�o��������)������̔X�E�B����邱�Ƃ��f�f����Ă���B
�A VHL�a�̉Ƒ������͂����肵�Ȃ��ꍇ
�����_�o�n���lj��邢�͖Ԗ����ǎ����(2�ȏ�)���� �����_�o�n���lj��܂��͖Ԗ����ǎ�ƈȉ��ɏq�ׂ�a�C������
�B ��`�q�����z��(��`�q�f�f��VHL��`�q�ُ킪�m�F���ꂽ�ꍇ)
����f�f��́A�Ƒ���������ꍇ�ƂȂ��ꍇ�ňقȂ�A�Ƒ���������ꍇ��VHL�a�ł݂���a�ς�1�ł��F�߂����VHL�a�Ɛf�f�ł��邪�A�Ƒ������Ȃ��ꍇ��VHL�a�ł݂����ᇂ��قȂ�2�ȏ�̑���ɑ��݂����VHL�a�Ɛf�f�����B�����_�o�n���邢�͖Ԗ��̑��������lj��͏]���AVHL�a�̐f�f��������ɂ͖������Ȃ��������A2003�N��Lonser��̕ȍ~�A���������lj������VHL�a�Ɛf�f����Ƃ����悤�ɕς���Ă��Ă���1, 2)�B����̐f�f���Lonser��̕ɏ������B���������lj��ʼnƑ������Ȃ��ꍇ�́A�����ɂ́A��`�q�f�f��VHL��`�q�ُ킪�m�F�����Ίm����VHL�a�Ɛf�f�ł���B
- Lonser R, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361�F2059-67.
- Hes FJ, Hoppener JW, LIPS CJ. Pheochromocytoma in von Hippel-Lindau disease. J Clin Endocrinol Metabol. 2003�G88�F969-74.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
�v��
���L�̕\����ʂɗՏ��I���ނƂ��ėp�����Ă���B
�\2 VHL�a�̕���
 (Lonser R, et al. Lancet. 2003�G361�F2059-67)1)
(Lonser R, et al. Lancet. 2003�G361�F2059-67)1)
|
������F�זE����������Ĕ��ǂ��Ȃ����A���ǂ��邩��VHL�a1�^(���F�זE��ǂȂ�)�AVHL�a2�^(���F�זE��ǂ���)�ƕ��ނ���B2�^�̂Ȃ��ł��t��ᇔ��ǂ̗L���ł����2�^A(�t��ᇂȂ�)�A2�^B(�t��ᇂ���)�ɕ��ނ��A����Ɋ��F�זE��݂̂����ǂ�����̂�2�^C�ƕ��ނ���B2�^�̂��̂̑�����VHL�`����Elongin C�ƌ������镔�ʂ̈ꕔ�̃A�~�m�_�ُ̈킪�����B�S�̂̂Ȃ���2�^�̐�߂銄����10�`20%�Ƃ�����B
- Lonser R, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361�F2059-67.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
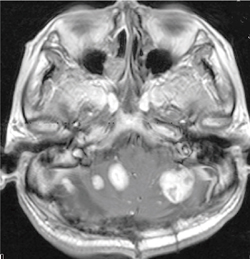
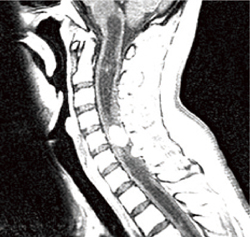
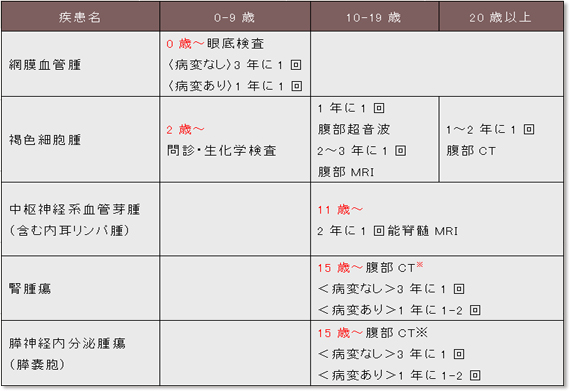
�����_�o���lj�� ���eMRI�ɂ������I�ȔZ�����ƔX�E�l�̏����Őf�f����(�}6-1a�A6-1b)�B
 |
 |
�}6-1a ���]���lj��
VHL�a�̑��������]���lj��B
�قڋψ�Œ����ȑ��e���ʂ�F�߂�B |
�}6-1b �ҒŌ��lj��
�Ґ��Ǘl�X�E�����Ґ����lj��B |
���������p�X�� ���eMRI(�ꍇ�ɂ�葢�eCT�lj�)�ɂĐf�f����(�}6-2)�B�����̒����_�o�n���lj��̐f�f�̍ۂɓ����ɍs���Ă������Ƃ��]�܂����B
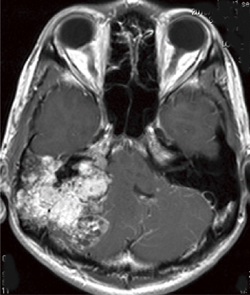 |
�}6-2 ���������p�X��
����������㓪�W�|���ɐi�W�����傫�ȓ��������p�X��B |
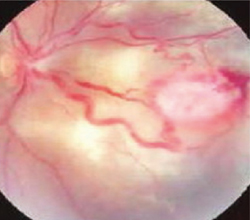
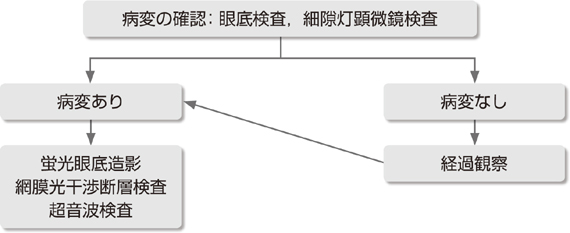
�Ԗ����ǎ� �U������ꌟ���A���������������ɂē����I�Ȍ��ǎ������(�}6-3a, 3b, 3c)�B
 |
|
| �}6-3a �Ԗ����ǎ� |
|
 |
 |
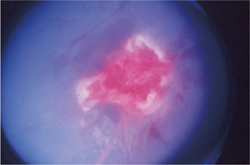
�}6-3b �Ԗ����ǎ�(���ÑO)
���ǎ���o����F�߂�B |
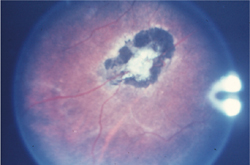
�}6-3c �Ԗ����ǎ�(���Ì�)
�Ԗ����ÌŔ���F�߂�B |
���F�זE��
(�X�N���[�j���O����)�����A���^�l�t�����E�m�����^�l�t����(Cr�)(������3�{�ȏ��z��) 24���Ԏ_���~�A�ɂ��B���^�l�t�����A�m�����^�l�t���������A�A�h���i�����A�m���A�h���i��������(��l�����3�{�ȏ��z��) �����J�e�R�[���A�~������(��l�����2�{�ȏ��z��)
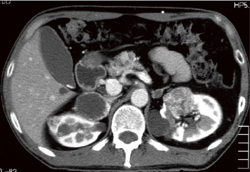
(�摜����)Dynamic CT(���eCT�̑�����)�A�P��MRI�ő������̓����I�Ȏ�ᇏ�����F�߂�(�}6-4a�A4b)�B
 |
 |
�}6-4a VHL�atype2B�ɔ��������E���t���F�זE��Ɨ����t���
1��̕��������CT�܂���MRI�Őf�f�\�B |
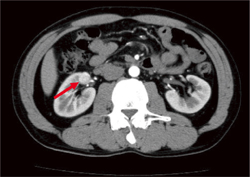
�}6-4b VHL�a�ɔ��������T�_�o�ߎ��(�p���K���O���I�[�})
�p���K���O���I�[�}(��)���w����艺��Ö������r���Ă���B |
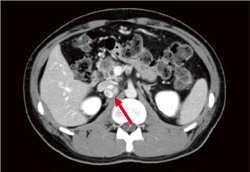
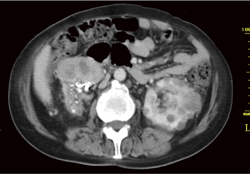
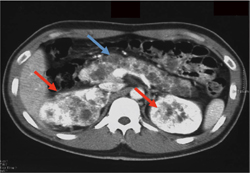
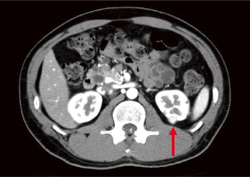
�t��� Dynamic CT(���eCT�̑�����)�A�P��MRI�ő������̓����I�Ȏ�ᇏ����������B�����Őt�X�E�̏�������������B����CT���X�X�E�A�X���̐_�o�������ᇂ��ɐf�f���邱�Ƃ��]�܂���(�}6-5a�A5b�A5c�A5d�A5e)�B
 |
 |
�}6-5a VHL�a�̗��t���
���t�̎�ᇂƍ��t�̔X�E��F�߂�B |
�}6-5b ���t��ᇂ��X�X�E�̍�����
��i���A�A���F�X�X�E�@�Ԗ��F�t��� |
 |
 |
�}6-5c �t��ᇏ���
���t�̔w���ɏ���ᇂ�F�߂�(��) |
�}6-5d �t��ᇏ���
�E�t�̕����ɏ���ᇂ�F�߂�(��) |
 |
|
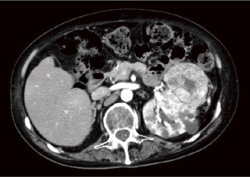
| �}6-5e ���t��ᇂƐt�X�E�̍����� |
|
�X�X�E �t��ᇂ�f�f����ۂ̑��eCT�A�����I�ȑ������X�E�̏���������(�}6-6a)
�X�_�o�������� Dynamic CT(���eCT�̑�����)�ŔZ�������ᇑ�������(�}6-6b)�B
�t��ᇂ̐f�f�̍ۂ̑��eCT�œ����ɐf�f���邱�Ƃ��]�܂����B
 |
 |
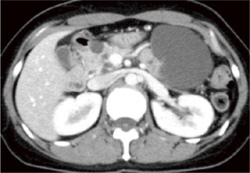
�}6-6a �X���ɑ召�̔X�E���a�ς��������Ă���B
(Tamura K, Nishimori I, Ito T, et al. Diagnosis and Management of pancreatic neuroendocrine tumor in von Hippel-Lindau disease. World J Gastroenterol. 2010�G6(36)�F4515-8���]��) |
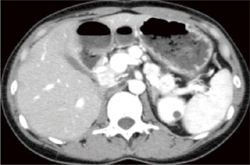
�}6-6b �X���ɑ��e�����ɔZ������鑽�����̎�ᇕa�ς�F�߂�B
(Maeda H, Nishimori I, Okabayashi T, et al.Total pancreatectomy for multiple neuroendocrine tumors of the pancreas in a patient with von Hippel-Lindau disease. Clin J Gastroenterol. 2009�G2�F222-5���]��) |
��������_�o�n���lj��ł͑��eMRI(Cr�l��1.5���Ȃ��ꍇ)�����������B���������p�X��͑��eMRI�A���eCT�����������B���������p�X��͒����_�o�n���lj��̐f�f���ɓ����ɍs���Δ픘���Ô�̖��ʂ�h�����Ƃ��ł���B�Ԗ����ǎ�ł͎U������ꌟ���ɂ���ᇂ̌����A���������������ɂ��u�h�E������Γ���Ȃǂ̍����ǂ̗L�����m�F�����������B���F�זE��ł́A�@�A�����^�l�t�����܂��̓m�����^�l�t�����A�A�A���A�h���i�����܂��̓m���A�h���i�����A��������l�����3�{�ȏ��z���Ƃ���B�����J�e�R�[���A�~���A�܂����ʐf�f�Ƃ��Ă͒P��T2MRI�AMIBG�V���`�O���t�B�[�ADynamic CT(���eCT�̑�����)���L�p�ł��邪�A���eCT�ł͍���������̗U���ɒ��ӂ��K�v�ł���B�܂������V�����^�l�t���������́A���F�זE��̐f�f�Ŋ��x�A���ٓx�Ƃ������A�ߓ����ɕی����ڗ\��ł���B�t��ᇂł�Dynamic CT(���eCT�̑�����)�A���������e�܃A�����M�[�A�t�@�\��Q�Ȃǂő��eCT���ł��Ȃ��ꍇ�͒P��MRI�����������B�X�_�o�������ᇂł�Dynamic CT(���eCT�̑�����)�����������B���̂��߂����̌����͐t��ᇂ̐f�f���ɓ����ɍs���Δ픘���Ô�̖��ʂ�h�����Ƃ��ł���B�����̏ڍׂ͊e��ᇂ̐f�f���Îw�j����ьo�ߊώ@�w�j���Q�l�ɂ��Ă������������B
�v��
| ����z���͖@(DNA�V�[�N�G���V���O)�ƌ���/�d�����o�@(��1)�ɂĖ�84%�Őf�f�ł���(�������A�����͌��݁A�ی��K���͂Ȃ�)�B |
�y��1�z����/�d�����o�@�F��ʓISouthern�AFISH�Aquantitative PCR�Areal-time PCR�Amultiplex ligation dependent probe amplification(MLPA)�Aarray CGH�@�Ȃ�DNA�̑�K�͂ȕψق����o������@
�����`�w�I�����Ɋւ���K�C�h���C���Ȃǂɂ��Δ��a����100%�̎����ł���A�\�h�@���Ö@���m�����Ă���A���Âɂ����QOL���ۂ���鎾���͈�`�q�f�f���s�����Ƃ��ł��鎾���Ƃ����B������VHL�a�͈�`�q�f�f�ŗ\������P���鎾���ł���ƍl������1)�B
��`�q�f�f�Ɋւ���葱�����ȒP�ɏq�ׂ�ƁA�Ώێ҂Ɏ����̓��e�ɂ��ď\���Ȉ�`�J�E���Z�����O���s���A��`�q�f�f�̖ړI�A���@�A�����҂ւ̉e�����܂ߗ\�z����錋�ʁA�������x(�����̌��E)�Ȃǂ��킩��₷���������������ŁA�팟�҂̈ӎu�ɂ�菑�ʂ̓��ӂčs���B�����N�҂̏ꍇ�͐e���҂̑���ɂ���čs���B���ʁA�J���̍ۂ͑Ώێ҂̈ӎu�Œm�錠���ƒm��Ȃ��ł��錠����ۏႳ��Ă���B���݁A�ی��K���͂Ȃ��s���Ă���{�݂͍��m��w��w����A��Ȃ݂̂ł���B
VHL��`�q�̖|��̈��639����(213�A�~�m�_)�ł��邪�Asplice���ʂُ̈�A3'���ُ̈��A��K�͂�DNA���̌����Ȃǂ����݂���B�ߋ��̉�͌��ʂł�VHL�a�̖{�M�ɂ������`�q�f�f�̐f�f���͖{�M��ł�84%�ł���2)�B�f�f���ʂ̓���͉���z���͂őΏێ҂�75%���f�f�\�ł���A����ɖ�9%��MLPA�@�Ȃǂ̌���/�d�����o�@�ɂ��DNA�̑�K�͂ȕψق��f�f�\�ł���3, 4)�B
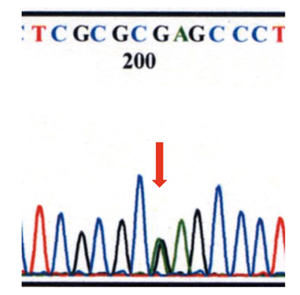 |
�}6-7 �_�C���N�g�V�[�N�G���X���
VHL��`�qexon1��208�Ԗڂ�G(�O�A�j��)��A(�A�f�j��)�ɒu�����A�R�h��70��Glu(�O���^�~���_)��Lys(���W��)�ɕψق����~�X�Z���X�ψق��݂���B |
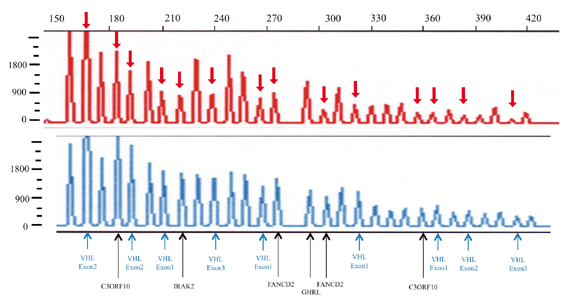 |
�}6-8�@Multiplex ligation-dependent probe amplification(MLPA)
| ��F |
���Ҍ��́GVHL��`�q�S�����A�Ԃ̖��(��)�̕���������R���g���[�����Ⴂ�B
���Ҍ��̂ł�PCR�����Y���ʂ�����R���g���[���ɔ�ׂĖ����x(65%�ȉ�)�Ɍ������Ă���B
3�Ԑ��F�̒Z�r���FANCD2��`�q�|C3ORF10��`�q�|VHL��`�q�|IRAK2��`�q�A�L�͈͂�DNA�f�Ђ̌�����B |
| ���F |
����R���g���[�� |
|
- ��`��w�֘A�̊w�(10�w���ь�����)�B��`�w�I�����Ɋւ���K�C�h���C��.2003.���{�l�ވ�`�w��Bhttp://jshg.jp/resources/data/10academies.pdf
- ������A�����\�B
- Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002�G30(12)�Fe57.
- Huang JS, Huang CJ, Chen SK, et al. Associations between VHL genotype and clinical phenotype in familial von Hippel-Lindau disease. Euro J Clin Invest. 2007�G37�F492.500.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
�v��VHL�a�͏���F�̗D����`�������ł��邽�߁AVHL�a���҂�f�f���Â��A�o�ߊώ@���s���ۂ͈�`�������Ƃ��Ĉ�`�q�f�f�ƈ�`�J�E���Z�����O���s���A�K�ȑΉ����Ƃ邱�Ƃ��]�܂��B
���
1.��`�������ɑ����`�J�E���Z�����O�̕K�v��
��`�J�E���Z�����O�ł͊��҂ƉƑ����K�v�Ƃ����`�w�I���Ƃ��ׂĂ̊֘A������A���ҁE�Ƒ��̃j�[�Y�𗝉����������ŐS���I�s������菜���A���Ȍ��肪�ł���悤�Ɏx������s�ׂł���B�q�g�Q�m���E��`�q��͌����Ɋւ���ϗ��w�j�Ɉ�`�������̌����̍ۂɂ͈�`�J�E���Z�����O���s�����Ƃ���������Ă���1, 3)�BVHL�a�͏���F�̗D����`�������ŁA�����ɓ��L�ȏǏ�������߁A���҂���Ɉ�`�q�f�f��f�f�A���ÂɊւ���L�p�ȏ�����Ĉ�`�J�E���Z�����O���s���x�����邱�Ƃ��K�v�ƂȂ�2)�B���݁A������w�t���a�@�Ȃǎ�Ȃ��_�a�@�ɂ͈�`���k���s����̐�������A���{�l�ވ�`�w��F���`���ォ��t�ȊO�̔F���`�J�E���Z���[�ɂ��J�E���Z�����O���s�����Ƃ��ł���B
2.��`�J�E���Z�����O�̉ߒ��Ɠ��e
- �a���̒����Ɖƌn�}�̍쐬
VHL�a�̏ꍇ�͒����_�o�n���lj��A�Ԗ����ǎ�A�t��ᇁA�t�X�E�A�X�X�E�A�X���(�_�o��������)�A������̔X�E�A�܂�ɔ畆�̌��ǎ�Ȃǂ����邱�Ƃɗ��ӂ��čs���B��1�x�ߐe�҂���A��2�x�ߐe�ҁA��3�x�ߐe�҂܂ł̌����҂ɂ��Đ��A���N�����A������(���Ǘ�)�A�N��A����(����)�Ȃǂ悷��3, 4, 5)�B
VHL�a�̈�`�q�������s���ۂ̐�������
VHL�a�ɂ��ċ�̓I�Ȑ���(����F�̗D����`�������A�Z����100%) �����ړI�ƌ������@�̋�̓I���� ��`�q�����̕��@�Ɛ��������ʂ��o��m��
���m��w��w����A��Ȃʼn���z���͂�MLPA�@�ŕ����Ė�84%�̊m���ł���B �\�z����闘�v ���z���̌��ʂ�����ꂽ�Ƃ���
�z���Ɗm�肵�ĕs�m��������̕s�������� ���ǂ̃��X�N��\���ł��� �\�h�I�[�u(�։������̖̉���A���N�f�f�̎�f�Ȃ�)��I���ł��A�����f�f�ɖ𗧂��A�l�X�ȍ����ǂɑ��A�����ɑΉ��ł��� ��`�q�̕ψق̏ꏊ���킩��A�Ƒ��̕��̈�`�q�f�f�ɖ𗧂�
���A���̌��ʂ�����ꂽ�Ƃ���
�\�z����郊�X�N�ƕs���v ���z���̌��ʂ�����ꂽ�Ƃ���
���A���̌��ʂ�����ꂽ�Ƃ���
�A���̌��ʂ��o�Ă�VHL�a�̉\���͊��S�ɔے�ł��Ȃ� ��L�̂���VHL��`�q������ł����ǂ̃��X�N������ꍇ������ �Ƒ����ɗz���҂�����ꍇ�A��肪������\��������
�������s��Ȃ����Ƃ̗��_�A���_ �����N�҂ł�15�܂ł͐e���҂̓��ӂ݂̂ōs���� 16�Έȏ�ł͐e���҂ƂƂ��ɑΏێ҂̓��ӂ��K�v�ƂȂ�
�v���C�o�V�[�̕ی� �������邱�Ƃ̎��R ��L(a)-(h)�̍��ڂɂ��Đ������ē���(informed consent)��B
��`�I���X�N�̐���ƕ]��(�Ĕ����ƐZ����)
��`�q�����̌��ʂ̐��� ��`�q�����̌��ʂ�(a)-(d)�ɊY������B
�a�I��`�q�ψ� VHL�a�̏ꍇ�͕a�C�ǂ��邱�Ƃ��m�肷���`�q�ُ킪����z���́AMLPA�@(��K�͂̈�`�q��������͂�����@)�ɂ���84%�Ŋm��ł���B�܂��AVHL�a1�^(Pheo�|)�AVHL�a2�^(Pheo�{)���ߋ��̕����I�Ȍ��ʂ��琄��ł���B
����I�Տ������ő����f�f���\�ł���B
�a�I���ǂ������f������ȕψ� SNP(��`�q���^�ɂ��ꉖ��u��)�ȂǂƔ��ʂ�����ȕψق����ɑ��݂���B�Ƒ����̊����NJ��҂̈�`�q�����̌��ʂ�萄�肷��B
�a�I�ȈӋ`�̂Ȃ��ψ� �ُ킪�F�߂��Ȃ��ꍇ ���̏ꍇ�͗Տ��I�f�f�ɗ��邱�ƂƂȂ�B
��`�q������̃t�H���[�A�b�v ������́A�K�X�J�E���Z�����O���p������B
3.��`�J�E���Z�����O�̕��@
��`�J�E���Z�����O�ł͔�w���I�Ή��A�����I�����A��e�I�ԓx�̃J�E���Z�����O��3���������K�v������B��`�J�E���Z�����O�ł͐��m�Ȉ�`��w�̒m�����킩��₷���`���邱�Ƃɂ��A��`�I���ŔY�ފ��҉Ƒ��̕s������菜���A���k�҂̍l�����A���A���O�̒m���A����́A�s���̑傫���A��Âɑ���M�������X�ňقȂ邱�Ƃɒ��ӂ���B
4.��`�J�E���Z�����O�̍ۂɍl�����ׂ�����
��`�J�E���Z�����O�́A�\�Ŏ��Əꏊ���߂čs���A���O�Ɍ����W�̂킩��ƌn�}����������悤���҂ɓ`���邱�Ƃ��]�܂��4, 5)�B
�@�����̈�`�a�ŁA���̃T�|�[�g�O���[�v����������Ă���B���҂̊�]��������̃O���[�v�ɘA�����Ƃ邱�Ƃ����߂Ă��悢�B��Îґ�����͓����Ȃ����A���҂͎��������ł͂Ȃ��Ƃ������g����������BVHL�a�ł́A�ق���Chain(http://www.vhl-japan.org/)�Ƃ������҉���݂���B
5.���̑��J�E���Z�����O���s���ۂɒ��ӂ��ׂ�����
�A�E�A�����A�D�P�ɂ��Ă����ӂ��B
������p�����Ĉ�`�J�E���Z�����O���ł��邱�Ƃ�ۏႷ��B
- �q�g�Q�m���E��`�q��͌����Ɋւ���ϗ��w�j(����16�N12��28���S������)
- ��`��w�֘A�̊w�(10�w���ь�����)�D��`�w�I�����Ɋւ���K�C�h���C��.2003.���{�l�ވ�`�w��z�[���y�[�W�Dhttp://jshg.jp/resources/data/10academies.pdf
- �M�B��w��w�������a�@��`�q�f�Õ��҃z�[���y�[�W(Genotopia)
http://genetopia.md.shinshu-u.ac.jp/genetopia/basic/basic2.htm
- ���Y�C�g��疾�C���c�^��DVon Hippel-Lindau�a�D�F�s�{����C�ďC�D�P���R�I�q�C����ېm�C���Ԍb�q�C���C�ҁD�Ƒ�����ᇈ�`�J�E���Z�����O�����_�Ǝ��ۄ��D�����F�����o�ŁG2000.p.297-301.
- �����`���D����̍s����`�J�E���Z�����O�D�F�s�{����C�ďC�D�P���R�I�q�C����ېm�C���Ԍb�q�C���C�ҁD�Ƒ�����ᇈ�`�J�E���Z�����O�����_�Ǝ��ۄ��D�����F�����o�ŁG2000.p.130-6.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 8 |
�e��ᇂ̌o�ߊώ@�Ǝ��ÃK�C�h���C�� |
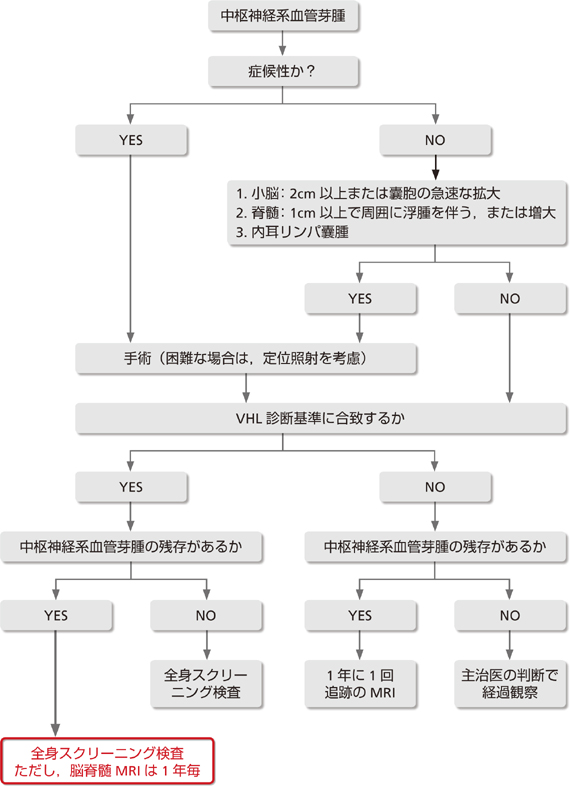
���̍��ŁA�e��ᇂ̐f�f���ÁA�o�ߊώ@�ɂ��ďq�ׂ�B�ߋ��̌o������VHL�a�͗c��������蔭�ǂ��AVHL�a�̉ƌn���Ŗ����ǂ̎҂�A��`�q�f�f�ɂ���Ă��łɐ��ݐ����҂Ɛf�f���Ă���҂̏ꍇ�͔��ǑO�ɂ���N���CT�AMRI�Ȃǂ̌��������I�ɎČo�ߊώ@���s�����Ƃ��K�v�ł���B�܂��A���Â��Ă������̎�ᇂ͍Ĕ����ł��肳��Ɍo�ߊώ@���s���K�v������B���̓_���炢�����̎�ᇂł͓��ɔ��ǑO�̐f�f���܂ށu�o�ߊώ@�v�Ɓu�f�f�Ǝ��Áv�Ƃ���2���ڂɕ����ďq�ׂ�B�܂��A�����_�o�n���lj��̍��ł́u���ː����Áv�����ʂȎ��Í��ڂƂ��Ĉʒu�Â����Ă���A���̕]�����q�ׂ�K�v������ƍl����ꂽ�̂ŕʍ��ڂƂ��ē��ꂽ�B
1.�o�ߊώ@
�v��
|
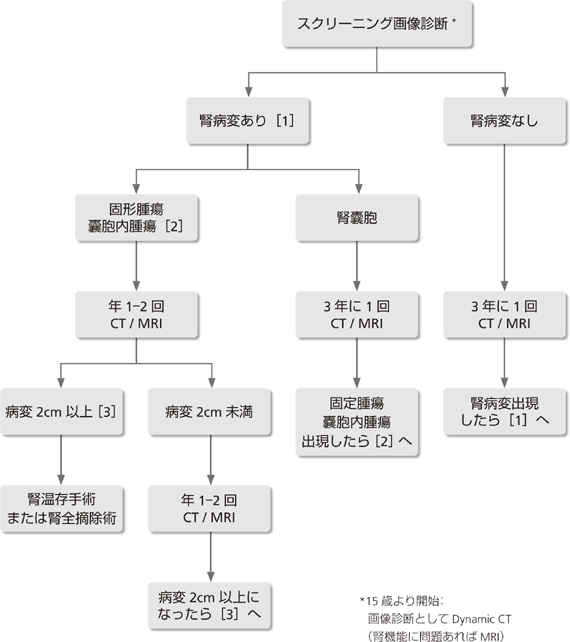
�n�C���X�N�Q(��`�q�����z����A�܂��͉Ƒ���������ꍇ�A������̔��ǂ�VHL�a�Ɛf�f���ꂽ�ꍇ)��11���2�N���ɑ��eMRI�������s���B
���]�ȂǁF2cm�ȉ��A�Ґ�1cm�ȉ��̖��nj�ᇂł��X�E���ᇎ��͂ɕ�����ꍇ�͋}���ɑ��傷��\��������̂�1)�A���N�`1�N��1��̌o�ߊώ@���s���B
|
���
Lonser��̕�1)�ɂ��ƁA�]�Ґ����lj��̕���(�͈�)���ǔN��́A���ʕʂł��ꂼ��A���]33(9�`78)�A�]��32(12�`46)�A�Ґ�33(12�`66)�ł���B���̃f�[�^�����ƂɁA���̑���a�ς̔��ǂ���s���AVHL�a�Ɛf�f���ꂽ�ꍇ�́A11����]�Ґ�MRI(���eT1�AT1�AT2�AFlair��)��2�N��1��s���B�nj�ᇂ������͖��nj��(���]�F2cm�ȏ�A�Ґ�1cm�ȏ�)���������ꂽ���_�œE�o�p�������͒�ʏƎ˂��s���B���̃T�C�Y�ȉ��̖��nj�ᇂł��X�E���ᇎ��͂ɕ�����ꍇ�͋}���ɑ��傷��\��������̂�2)�A���N�`1�N��1��̌o�ߊώ@���s���B
2.�O�ȓI����
�v��
|
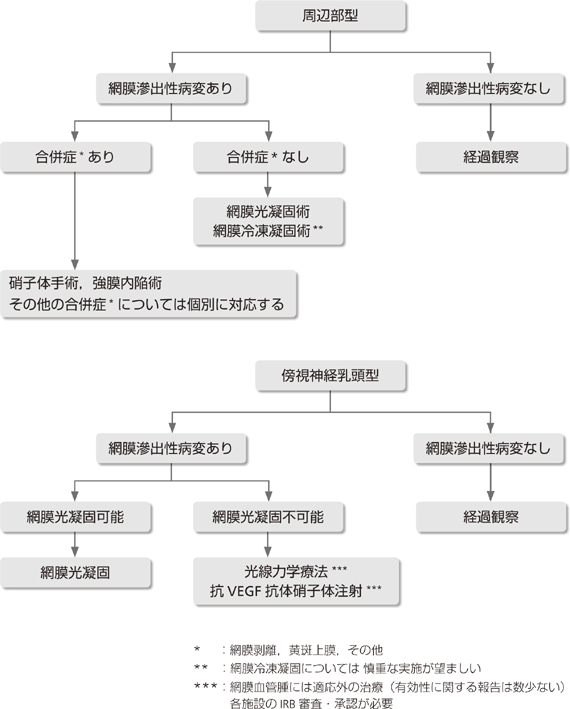
�����_�o�n�̌��lj��͏nĵ��͔̂]���[��������ᇂ������Ď�p�E�o���s���B
���nj�ᇂɂ͌����I�ɂ͏njƂȂ����Ƃ��ɍs�����Ґ���ᇂł�1cm�ȏ�A�܂��͑���X����������͖̂��Ǐ�ł���p�����������B
|
���
�����_�o�n�̌��lj��͎�ɏ��]�A�]���A�Ґ��ɔ�������BMRI�ɂĎ�ᇂ��m�F���ꂽ�nĵ��͔̂]���[��������ᇂ̏ꍇ�������Ċ�{�I�Ɏ�ᇓE�o�p�����������B�Ґ���ᇂƔ]������ᇂ͏Ǐi�s����ƓE�o���s���Ă��Ǐ�̒����ȉ��P�����Ȃ����Ƃ���A�Ǐy�x�ł��邤���ɓE�o��p���l������B�������̎�ᇂ͑S�E�o���s���A�X�E����ᇂ́A�Ǎ��߂̂ݓE�o����B
���nj̎�ᇂɊւ��ẮA��ᇎ����E�X�E�Ƃ��Ɉ�葬�x�ő��傷��Ƃ͌��炸�A���鎞���ɋ}���ɑ��傷�邱�Ƃ�����̂Œ���I��MRI�������p�����邱�Ƃ��d�v�ł���B�܂��X�E�͎�����ᇂ������呬�x���������ߒ��ӂ��K�v�ł���B
���]�̖��nj�ᇂ͏njɂȂ��Ă����p���s�����Ƃ������Ƃ��邪�A1)���a��2cm�ȏ�A2)�摜���ᇂ܂��͔X�E�̋}���Ɋg����݂����͖̂��njł����Ă���p�ɂ��E�o�����������B
�Ґ���ᇂ͖��njł��A1)1cm�ȏ�A2)��ᇂ̎��ӂɕ�������́A�܂���3)����I��MRI�ɂ���ᇂ܂��͔X�E�̑��傪�݂�����͓̂E�o���s��9)�B
�]������ᇂ́A�nj܂���1cm�ȏ�̖��nĵ��̂ŁA�Ȃ����]���\�ʂɈʒu������̂͑�����p�ɂ��E�o�p���l������B�]���[���ɑ��݂�����͎̂�p�ɂ��E�o������Ȃ��̂�������ː����Â��l������(��9�� �t���[�`���[�g�Q��)�B
3.���ː�����
�v��
|
�O�Ȏ�p������ȏꍇ�A��ʕ��ː����Â��l�������B
�Ґ��E�]���������̂��̂��܂߂Č��ʂ͊��҂����B
���nja�ςɑ���\�h�I�Ǝ˂͊��߂��Ȃ��B
��ᇐ��䗦�͎��Ì�5 �N��8���قǂł���B
�g�傷��X�E�ɂ͓K�Ȏ��Ö@�ł͂Ȃ��B
|
���
���ː����ẤA�njƂȂ������̂��邢�͑���X���𑱂����ᇂɑ��āA��p�E�o���X�N�������K���łȂ��Ɣ��f����鎞�ɑ�2�I�����Ƃ��ėp������B������̕�ł͒�ʕ��ː����Âɂ���ᇐ��䗦(���Ȃ��Ƃ���ᇂ����債�Ȃ�)�͎��Ì�5�N��80%�ȏ�ł���B2009�N��Moss��̕ł́A��ʕ��ː����Â����ꂽ31��82�a�ςł̋Ǐ����䗦�́A3�N��85%�A5�N��82%�ł������Ƃ����B5��ŕ��ː���(���ώ�ᇕӉ�����28.2Gy)���������A���̂�����2�Ⴊ�njł������B����A�ƎˑO�ɏnjł�����41�a�ϒ���36�a��(88%)�ŗՏ��Ǐ��P�����B������2010�N��Asthagiri��͎��Ì�5�N�̎�ᇐ��䗦��Moss��̕��l83%�ł��邪�A10�N�ł�61%�A15�N�ł�51%�܂Œቺ���邱�Ƃ���Ă���B�Ґ����lj��̎��Ð��т͓��W���a�ςƓ��l�ł���B�g�傷��X�E���������ƂȂ��ɂ͕��ː����Â̑I���͓K�ł͂Ȃ��BVHL���L�̖��́A��ʕ��ː����Â�������ᇂɋߐڂ������ʂɐV���Ɏ�ᇂ����債���ꍇ�ɁA�lj��̏Ǝ˂ł͕��ː��Ǝ˖�̏d���������邱�Ƃł���B
�v��
|
�o�ߊώ@�F11����2�N���ɒ����_�o�n���lj��X�N���[�j���O�Ɠ�����MRI�ƒ��͌����ɂĐf�f����B
�f�f�Ǝ��ÁF�f�f�́A�Ǐ�E���͌����E�摜�f�f(MRI, CT)�ɂ���čs���A�������ꂽ�ꍇ�͒��͒ቺ�ɒ��ӂ��Ȃ���ϋɓI�Ɏ�p���s���B
|
1.�o�ߊώ@
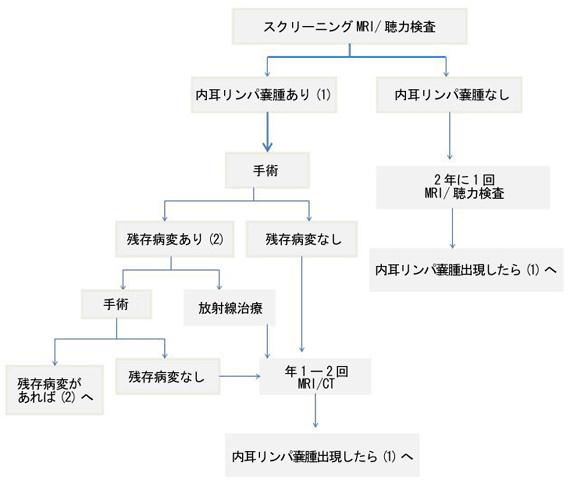
VHL�a�ɔ������������p�X��́A���Ăł�VHL�a���҂�11�`16%�ɔ��ǂ��邱�Ƃ�����23-25)�A���������p�X��S�̂�5�`15%��VHL�a�ɔ������̂Ƃ����25)�BVHL�a�̏ꍇ�A���������p�X��̔��ǎ����́A11�`63�ŕ���22���ƍl�����邽��24,26)�A���������p�X��̃X�N���[�j���O�́A11����2�N���ɒ����_�o�n���lj��̃X�N���[�j���O�Ɠ����ɒ��͌���(audiometry)�Ɠ����̍��𑜓xMRI�iT1�������A���eT1�������AT2�������AFlair���j���s��3)�B�X�N���[�j���O�œ��������p�X��F�߂���ΐϋɓI�Ɏ�p���s�����Ƃ��E�߂���27-29)�B��p���s���Ď�ᇂ��c�����Ă���ꍇ�͍Ď�p���s�����A���ː����Â��s���āA���N�`1�N���ɒ��͌����AMRI�ACT�ɂăt�H���[�A�b�v���s���B�܂��Б��̓��������p�X���VHL�a���҂̖�30%�͑Α��̓��������p�X��������ꔭ�ǂ��邽��29)�A�Б��̓��������p�X������Â����ꍇ�́A�Α����܂�1�N����MRI�A���͌����ɂČo�ߊώ@����B
2.�f�f�Ǝ���
���������p�X��̐f�f�́A�Տ��Ǐ�A�摜�f�f�A���͌���(audiometry)�ɂ��s���B�Տ��Ǐ�́A���͒ቺ(95%)�A����(92%)�A῝�(62%)�A����(29%)�A��ʐ_�o���(8%)��悷��29)�B�摜�f�f�ł͕`�o����Ȃ������ȓ��������p�X������݂��邪30)�A���������p�X��摜�f�f�ŕ`�o����Ă���ꍇ�́A����MRI�̑��eT1�������ő��e�a�ςƂ��ĕ`�o���� (100%)�A�����ȓ��������p�X��͓���MRI��Flair���Ŗ����H���̌�����������鍂�M���Ƃ��ĕ`�o�����(38%)28)�B�����̍��𑜓xCT�ł͓������p�ǂ̍��Z�𑜂Ƃ��ĕ`�o�����27)�B���͌����ł́A�i�K�I�Ȓ��͒ቺ(43%)�܂��͋}���i�s���̒��͒ቺ(43%)��F�߁A�ɏ��i�s���̒��͒ቺ(14%)�͏��Ȃ�29)�B�����̓��������p�X��ł́A��r�I�ቹ�悩�璮�͂���Q�����X��������29, 30)�B
���������p�X��̎��ẤA�����Ƃ��ĊO�ȓI�؏��ł���27-29)�B��ᇂ͏������Ă��ˑR���͏��������������Ƃ�����̂Ŕ������ꂽ�玡�Â𑁊��Ɍv�悷��27, 29)�B��x���͂��������͔̉��ɍ���ł���A�����i�K�ŏǏ���Ď����T�d�ɑΏ����邱�Ƃ͒��o���������邽�߂ɂ͏d�v�ł���B��p���@�́A��ᇂ̑傫���ɂ���ĈقȂ邪�A�����̏ꍇ(73%)����H���̐؏��@(retrolabyrinthine petrosectomy)�ɂ��s���27, 28)�A��ᇂ�E�o����B��ᇂ����������������p�X���ɗ��܂��Ă���Β��͂̉������\�ł���27)�B�ʂ̎�p�A�v���[�`�Ƃ��Čo���@(12%), �O/��S��Ö����@(9%), �o���H���邢�͑�����/�o���H�@(6%)�ōs����ꍇ������28)�B�Տo�����ő傫�Ȏ�ᇂł͏p�O�ɍǐ��p���L���ȏꍇ������28)�B�p��̏Ǐ�ł́A���͂͏p��97%�ŕs�ς����P���A�ቺ��3%�ł������B῝�͏p��86%�ŏ������A14%�ʼn��P�����B���́A�p��96%�ŏ����������A4%�ł͕s�ςł������B������ �p��77%�ŏ����������A23%�ł͕s�ςł������B��ʐ_�o��Ⴢ͏p��75%�ʼn��P������25%�ł͕s�ςł�����28)�B���ː����Â͎�ᇂ��c�������ꍇ�ɍs���邱�Ƃ�����A���̌��ʂ͒�܂��Ă��Ȃ����̗̂L�p������������Ă���28)�B����(��9�̓t���[�`���[�g�Q��)�B
�v��
�V�������o�ߊώ@���J�n����B
��ꌟ���ɂ��f�f���邪�A�u����ꑢ�e�����Ȃǂ̕⏕�������d�v�ł���B
���Â̊�{�͖Ԗ����Ìłł��荇���ǂɑ��Ď�p���s���B
�@�T���_�o�����^�ł͖Ԗ����Ìł��s�\�ȏꍇ������B
|
���
1.�o�ߊώ@31, 32)
�V�����Ŋ�ꌟ�����s���B���a�ς�F�߂Ȃ��ꍇ�A3�N���Ɋώ@����B�a�ς�F�ߎ��͂ɉe�����y�ڂ��ꍇ�͓K�X�A�e�����y�ڂ��\���̒Ⴂ�ꍇ��1�N���Ɋώ@����B
������a�ς�F�߂����ߊ�ꌟ�����s���ꍇ�́A�a�ς�F�߂Ȃ����2�N���Ɋώ@����B�a�ς�F�ߎ��͂ɉe�����y�ڂ��ꍇ�͓K�X�A���͂ɉe�����y�ڂ��\���̒Ⴂ�ꍇ��1�N���Ɋώ@����B
2.����31, 32)
��ꌟ���ƍ��������������ɂ��f�f����B�a�ς�F�߂�ꍇ�͌u����ꑢ�e�����A�Ԗ������f�w�����A�����g�������s���B
3.����
���ӕ��^ �Ԗ����o���a�ς�����ΖԖ����Ìł��s��33)�B�Ԗ����Ìłɂ͕a�����NjÌłƉh�{���NjÌł�2��ނ�����B�����ǂɑ��Ă͋������p��Ɏq�̎�p���s���B�Ԗ��Ⓚ�Ìłɂ��Ă͐T�d�Ȏ��{���]�܂����B
�T���_�o�����^ �Ԗ����o���a�ς�F�ߖԖ����Ìʼn\�ȏꍇ�͖Ԗ����Ìł��s��33)�B�s�\�ȏꍇ�̎��Ö@�͊m������Ă��Ȃ��B�����w�I���Ɏq�̒���34)������͊w�Ö@35)�̌��ʂ�����Ă��邪�A�e�{�݂̗ϗ��ψ���ŐR������K�v������(��9�� �t���[�`���[�g�Q��)�B
�v��
|
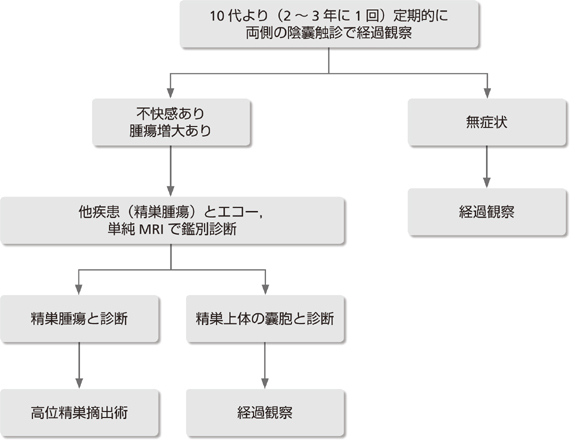
VHL�a2�^�ƌn�ł́A2����f�ƔA�E���t�̃z�������������J�n�A10���摜�����������̕����a�ςƓ����ɃX�N���[�j���O���s���B
��p�ł͉\�Ȍ��蕛�t�����؏����s���A�玿�@�\�������͂���B�܂����o���Ȃǂ̒�N�P��Z�������߂���B
|
���
VHL�ł͊��F�זE��̔��ǂ��Ȃ��ƌn(1�^�ƌn�AVHL type1)�ƁA�D������ƌn(2�^�ƌn�Atype2)���m���Ă���A��҂ł�90%�ȏ�̊��҂Ŋ��F�זE��̔��ǂ��݂���ƌn������36, 37)�B���ǔN���3�Ƒ�������݂��邱�Ƃ�����B����AVHL��͈�ʗ���z�����������A�Տ��Ǐy�����̂�����38-42)�B
1.�o�ߊώ@
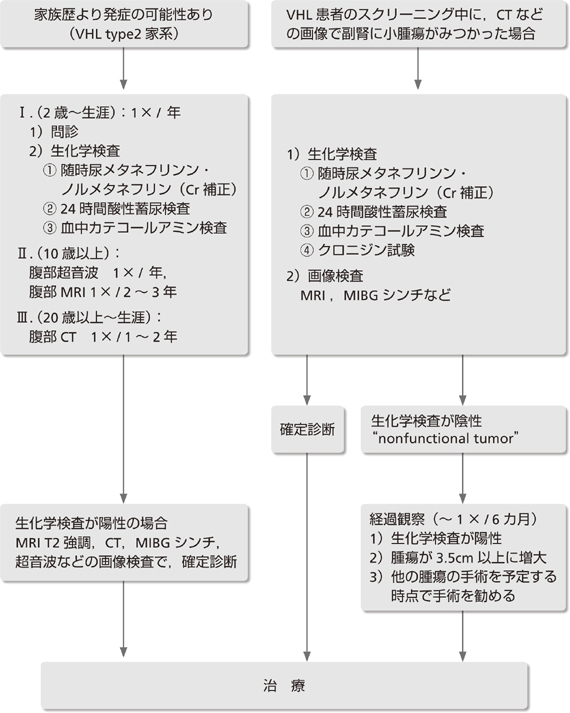
�Ƒ�����蔭�ǂ̉\��������ꍇ(VHL type2�ƌn)�B
(2�`���U)�F1�~/�N�ŁA
��f(���F�זE��ɓ��L�ȏǏ�̒���) �����w����
(�X�N���[�j���O����)�����A���^�l�t�������E�m�����^�l�t����(Cr�)(��l�����3�{�ȏ��z��) 24���Ԏ_���~�A�ɂ��A���^�l�t�����A�m�����^�l�t���������A�A�h���i�����A�m���A�h���i��������(��l�����3�{�ȏ��z��) �����J�e�R�[���A�~������(��l�����2�{�ȏ��z��)
(10�Έȏ�ʼn摜���������A���̕����a�ς������ɃX�N���[�j���O)�F ���������g1�~/�N�A����MRI 1�~/2�`3�N
(20�Έȏ�`���U)�F ����CT 1�~/1�`2�N
�Ȃ��AMIBG�V���`�͔팟�҂̕��S���傫���̂Ŋm��f�f�ɗp���A�ʏ�̃X�N���[�j���O�Ƃ��Ă͂����߂Ȃ��B
2.�f�f�Ǝ���
�f�f�͈�ʗ�̊��F�זE��Ɠ��l�ɍs���B����A�摜�����ŋ��R�݂������A�����Ȕ�@�\���̂��̂ł͌o�ߊώ@���\�ł���B���̏ꍇ�A�`1�~/6�J���̃t�H���[�������s���A1)�����w�������z�����A2)��ᇂ�3.5cm�ȏ�ɑ���A���邢�́A3)���̎�p��\�肷�鎞�_�Ŋ��F�זE��̎�p�������߂�43)�B���Â͒ʏ��Ɠ��l�ɐ؏���p���s���BVHL�ł͓������A�َ����ɑ������A������̎�p�̉\��������̂ŁA�\�Ȍ��蕔���؏��ɂ�蕛�t�玿�@�\�̉������͂���B�܂����o���Ȃǂ̒�N�P��Z�������߂���43, 44)�B
3.�ӕʐf�f
�ӕʐf�f�Ƃ��ẮAVHL�a�ȊO�̊��F�זE��ǂ����`�������A���Ȃ킿�������������(MEN)�A�_�o���ێ�LJT�^(NF1)(���b�N�����O�n�E�[���a)�A��`���T�_�o�ߎ�nj�Q(�R�n�N�_�E���f�y�f(Succinate Dehydrogenase�FSDH)�ψٗz���T�_�o�ߎ�)�Ȃǂ��l������B
�v��
�o�ߊώ@
�t��ᇐf�f�̂��߂̃X�N���[�j���O��15�ɊJ�n���A���U�ɂ킽��o�ߊώ@����B�f�f���@�Ƃ��Ă�Dynamic CT(���e����CT)�����������B
�f�f�Ǝ���
��ᇌa��2cm�����Ƃ���Ŏ��Â��l������B���Ö@�Ƃ��Ă͐t������p�����������B
�t�X�E�ɂ��ẮA�T�C�Y�ɂ�����炸�o�ߊώ@�����������B
|
���
1.�t��ᇂ̃X�N���[�j���O(�f�f)����ьo�ߊώ@
VHL�a�ɔ����t��ᇂ̔��ǎ�����15�ΑO��ƍl�����Ă��邽�߁A�t��ᇂ̃X�N���[�j���O��15�ɊJ�n����B�摜�f�f�@�Ƃ��ẮADynamic CT���ł��D��Ă��邪�A�t�@�\��Q������ꍇ��MRI��p����B���AVHL�a�ɔ����t��ᇂ�Birt-Hogg-Dube (BHD)�ɔ����t��ᇂ��������A�������Ƃ����ʂŗގ����邪�ADynamic CT�Ŋӕʉ\�ł���Ƃ���Ă���B
�o�ߊώ@���Ɏ�ᇐ��a�ς��m�F���ꂽ�ꍇ�A�N1�`2��摜�f�f���s���A��ᇌa��2cm�ɂȂ�܂Ōo�ߊώ@����B��ᇌa��2cm�ɂȂ����i�K�Őt�a�ςɑ��鎡�Â��l������B�t���Ɏ�ᇐ��a�ς�F�߂Ȃ��ꍇ�́A3�N���ɉ摜�f�f���s���B�t��ᇂ͐��U�ɂ킽���Ĕ��ǂ̃��X�N�����邽�߁A�o�ߊώ@�ɂ��Ă͐��U�ɂ킽��s���K�v������B�܂��A��ᇂ̈������̂��ߕs�ǂȗ\���悷�邱�Ƃ�����A���ӂ��K�v�ł���B
2.�t��ᇂ̎���
��ᇕa��(�Ō`��ᇂ���єX�E�����)��2cm�ȏ�ɂȂ������_�ŁA��ᇂɑ��鎡�Â����߂�B�ȑO�́A���ẴK�C�h���C���ɏ]��3cm����Ƃ��Ă������A�t������p���l�����ꍇ���a�Ŏ��Â��J�n�����ق����L���Ȃ��ƁA�ߔN�����Ö@�̂悤�Ȓ�N�P���Â��\�ɂȂ������Ƃ��l�����āA2cm���J�b�g�I�t�Ƃ��邱�ƂƂ���45)�B��p�̊�{�́A�t������p(�t�����؏��p�܂��͎�ᇓE�o�p)�ł��邪�A��ᇂ̑��ݕ���(���S��������Ȃ�)�A��ᇔ������̎�ᇌa���傫�����̂��ᇐ��������ł���Ȃǂ̗��R�Őt������p���Z�p�I�ɍ���ȏꍇ�͐t�S�E���p���I�������46)�B�܂��ߔN�{�݂ɂ���Ă͓������Â����{�\�ƂȂ��Ă���B�t�X�E�ɂ��ẮA�T�C�Y�ɂ�����炸�o�ߊώ@�����������B (��9�� �t���[�`���[�g�Q��)�B
1.�o�ߊώ@
�v��
|
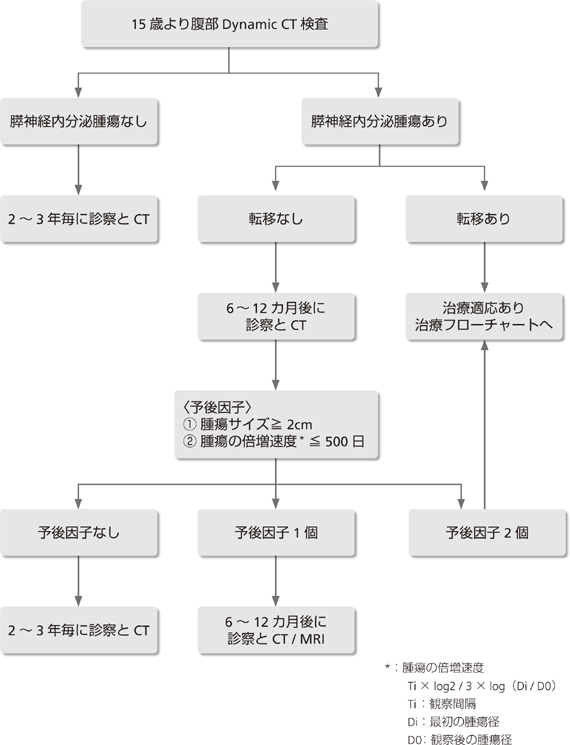
��I�ȕ�������̌o�ߊώ@�̈�Ƃ���15���Dynamic CT�������s���B
P-NET�̂Ȃ��ꍇ�A3�N���ɕ���Dynamic CT�������s���B
P-NET������A���u�]�ڂ̂Ȃ��ꍇ�͎��ÓK���ɂ��Č�������B
6�`12�J����ɕ���Dynamic CT���Č����A2�̗\����q(�@�ő��ᇃT�C�Y��2cm�A�A��ᇂ̔{�����x��500��)�̐��ɂ�莟��̌��������Ȃ�тɎ��ÓK�������肷��B
�\����q��0�F2�`3�N��ɕ���Dynamic CT�������s���B
�\����q��1�F6�`12�J����ɍēx����Dynamic CT�������s���B
�\����q��2�F�����s���B
P-NET������A���u�]�ڂ��Ă���ꍇ�A���Â��s���B
|
���
VHL�a��8�`17%�̏Ǘ�ɂ������X�_�o�����含���(pancreatic neuroendocrine tumor;P-NET)�̍������݂���47)�BVHL�a�ɍ�������P-NET�̂قƂ�ǂ͔�@�\���Ŗ��njł��邪47-49)�A��N��蕠���̃T�[�x�C�����X�������J�n����邽�߁A��ʂ̔�@�\��P-NET�ɔ�ב����ɔ�������邱�Ƃ�����50)�A�܂��A�f�f���ɉ��u�]�ڂ݂̂���Ǘ��11�`20%�Ə��Ȃ�50)�BVHL�a�̗L���ɂ�炸P-NET�̔���͈�ʂɊɏ��ł���BP-NET�����S�����ƂȂ�Ǘ��NIH(National Institutes of Health)�ɂ����錟���ɂ���52)�AVHL�a�S�̂�0.3%(����633��ł̌���)�AP-NET����������VHL�a��1.9%(����108��ł̌���)�ł���A�\��͔�r�I�ǍD�ł���48)�BVHL�a�ɂ�����P-NET�͊��F�זE��Ƃ̍����������X���ɂ��邪50, 51)�A����͓����Ă��Ȃ�52)�B
����܂�P-NET����������VHL�a�̍ŔN�����12��(����)��53)�A16�̕�48)�������B�t��ᇂɑ���T�[�x�C�����X��15����J�n����邱�ƁA���ː��픘�̉e���A���e�܂ɂ��t��Q�Ȃǂ��l�����A��I�ȕ�������̃T�[�x�C�����X�Ƃ���15��蕠��Dynamic CT�������J�n����(��9�� �o�ߊώ@�t���[�`���[�g�Q��)�B������P-NET�̕`�o���x��Dynamic CT�������ł��D��Ă��邪54)�A�����g������(EUS)���ł��D��Ă���Ƃ̕�����B�̓]�ڕa�ςł�MRI���L���Ȃ��Ƃ�����55)�B
����̕���CT�T�[�x�C�����X(15�Ύ�)�ɂ�����P-NET�̂Ȃ��ꍇ�́A3�N��(��)�̕���Dynamic CT���������������B�{�M�̍ŋ߂̉u�w�����ɂ��AP-NET�f�f���ɉ��u�]�ڂ��F�߂�ꂽ�Ǘ�́A7.5����VHL�a�̂Ȃ�P-NET���҂Ɣ�r�����Ȃ�56)�BP-NET������A���u�]�ڂ��Ă���Ǘ�͎��ÓK�p�ƂȂ�(���L�u�f�f�Ǝ��Áv���Q��)�BP-NET�̃T�[�x�C�����X�ɂ����Ė��ƂȂ�̂́AP-NET�����艓�u�]�ڂ̂Ȃ��Ǘ�̎�舵���ł���B��ʂ̔�@�\��P-NET�́A���ׂĎ�p�̓K���ƍl�����Ă���57, 58)�A�܂��T�^������������B��NJ��҂ɔ��ǂ�����@�\��P-NET���A�̓]�ڂ��F�߂���O�ɑ����Ɏ�p���ׂ��ƍl�����Ă���59)�B������VHL�a�ɂ�����P-NET�́A1)�������邢�͍Ĕ����������ƁA2)VHL�a�ł͐t��ᇍ���������Ȃ��Ȃ��A���ꂾ���ŕ�����̎�p���K�v�Ȃ��Ƃ����邱�Ƃ���P-NET��p�K���̌���ɂ͐T�d��v����B
P-NET����������VHL�a�̗\����q�Ƃ��āA�@�ő��ᇌa��3cm�A�AVHL��`�q�G�N�\��3�̕ψفA�B��ᇂ̔{�����x��500����3������Ă���48)�B�����3�̗\����q�̂Ȃ��ǗႠ�邢��1���q�݂̂�L����Ǘ�ł͉��u�]�ڂ��݂��Ȃ��̂ɑ��A2���q�����Ǘ�ł�33%�A3���q��L����Ǘ�ł�67%�ɉ��u�]�ڂ�������48)�B�������A��`�q�����͑S�Ǘ�Ŏ{�s����Ȃ����ƁA��`�q�ُ�̌��o����80%���x�ł��邱�Ƃ��60)�A�킪����VHL�a�T�[�x�C�����X�Ɉ�`�q�������ʂ��܂ނ͎̂��������ƍl������B
����A��ᇂ̍ő�a�A�{�����x�́A��p�K���f���邤���ŏd�v�Ȉ��q�ł���BP-NET�����艓�u�]�ڂ̂Ȃ��Ǘ�ł́A��ᇂ̑��B���x�̔���̂��߁A6�`12�J����ɍēx����Dynamic CT�������s���B���̍ہA��ᇌa��2cm�̏Ǘ�ł͂��Z�������Ԋu(6�J����)�A��ᇌa��2cm�̏Ǘ�ł�1�N��̍Č��������������B�Ȃ��A��L�̂悤�Ɉ�`�q�������ʂ�\����q���珜�O�������ƁA2cm�a��P-NET�ł����u�]�ڂ̂���ǗႪ���邱��48)�A��ʂ�(VHL�a�̂Ȃ�)��@�\��P-NET�ł͎�ᇌa�ɂ�����炸��p����������Ă��邱��57, 58)���l�����A�\����q�ɂ�����ő��ᇌa��2cm�ȏ�Ƃ����B
2��ڂ̃T�[�x�C�����XCT�����ɂ��2�̗\����q(�@�ő��ᇃT�C�Y��2cm�A�A��ᇂ̔{�����x��500��)�肵�A�o�ߊώ@�Ȃ�тɎ��ÓK�������肷��B���Ȃ킿�A2�̗\����q�̂Ȃ��Ǘ��2�`3�N��ɁA1���q�����Ǘ�ł�6�`12�J�����3��ڂ̃T�[�x�C�����XCT�������s��(��9�� �o�ߊώ@�t���[�`���[�g�Q��)�B����A2���q�Ƃ��z���̏Ǘ�͓]�ڂ̉\���������A���炩�̎��Â��K�v�ł���B
2.�f�f�Ǝ���
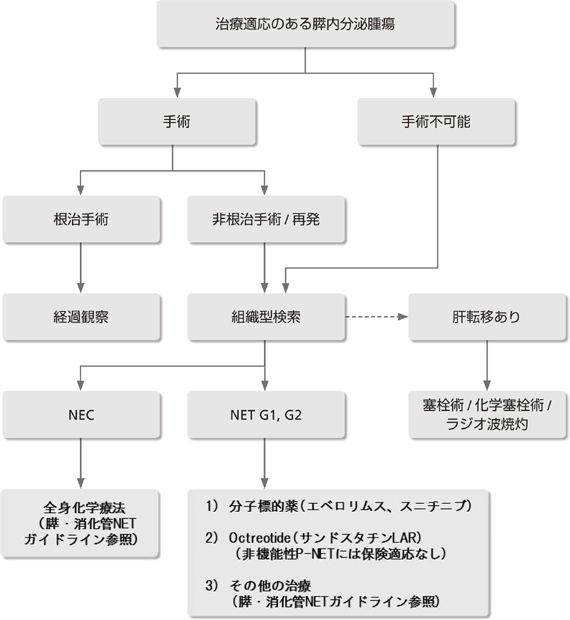
�v��
|
���ÓK������Ƃ����Ǘ�ɂ͈ȉ��̌����ɏ]�����Â��s���B
���u�]�ڂ̗L���ɂ�����炸�A�؏��\�ȏǗ�͎�p���s���B
��p�͎�ᇊj�o�p����{�Ƃ��A�\�Ȍ����X�@�\����������p�����l������B
��p�s�\�A����p�܂��͏p��ɍĔ������Ǘ�ł́A���Âɂ������ᇂ̕����x(WHO����)���l������B
NET G1�AG2�̏ꍇ�A���q�W�I��i�G�x�������X(�A�t�B�j�g�[��)�A�X�j�`�j�u(�X�[�e���g)�����������B
Neuroendocrine carcinoma(NEC)��VHL�a�ɍ������邱�Ƃ͋H�ł��邪�A���������ꍇ���X�E������NET�f�ÃK�C�h���C���ɏ����Ď��Â���B
�̓]�ڂ����݂���ꍇ�A�Ǐ��Ö@���l������B
|
���
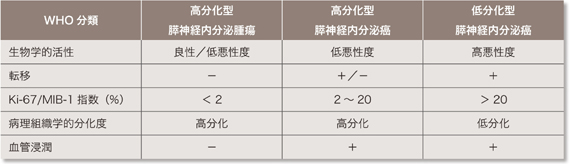
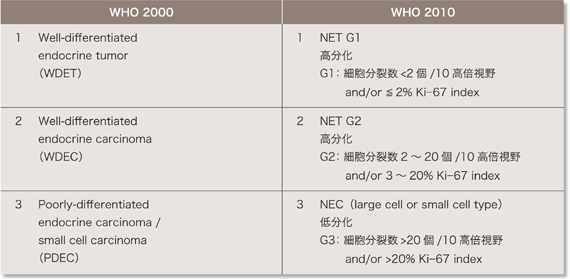
WHO�ł�P-NET���w�I�����A�]�ڂ̗L���AKi-67/MIB-1 �w���A�a���g�D�w�I�����x�A���ǂւ̐Z���A��ᇌa�Ɋ�Â��āA�������^�X�_�o�������ᇁA�������^�X�_�o��������A�ᕪ���^�X�_�o��������ɕ��ނ��Ă����B(�\1)61, 62)�BWHO ���ނ�2010�N�ɂ���ɉ�������ANET G1�AG2�ANEC(neuroendocrine carcinoma)�ƕ��ނ��ꂽ�B�����ނƂ̑Δ��\2 �Ɏ���63)�B�܂��AVHL�a�ɂ�����P-NET�͂قƂ�ǔ�@�\���ł���B�܂�VHL�a��NEC���������邱�Ƃ͔��ɏ��Ȃ��B��L�̃T�[�x�C�����X�ɂ�莡�ÓK������Ƃ����Ǘ�ɂ͈ȉ��̌����ɏ]�����Â��s��(��9�� ���Ãt���[�`���[�g�Q��)�B���Ȃ킿�A���u�]�ڂ̗L���ɂ�����炸�A�؏��\�Ȏ�ᇂ͊j�o�p���s���B��p�͎�ᇊj�o�p����{�Ƃ��A�\�Ȍ����X�@�\����������p�����l������B�܂��AP-NET �̎�p�ɍۂ��A���̕�������̍��������ɑ����p�������l������B��p�s�\�A����p�܂��͏p��ɍĔ������Ǘ�ł͑g�D�w�I�������s���B
2013�N�ɖ{�M�ł͂��߂āu�X�E�����ǐ_�o��������(NET)�f�ÃK�C�h���C���v�����\���ꂽ��64)�AWHO����2010�ɂ�����NET G1�AG2�̏ꍇ�A���q�W�I��̃G�x�������X(�A�t�B�j�g�[��)��X�j�`�j�u(�X�[�e���g)����������Ă���NEC�̍����͔��ɏ��Ȃ����A���̏ꍇ�K�C�h���C���ɏ]���ĉ��w�Ö@���s��64)�BOctreotide(�T���h�X�^�`��LAR)���^���؏��s�\�̍������^��@�\���̒����_�o�������ᇂ̗\������P�����Ƃ̕�����(PROMID����)65)���A��@�\��P-NET�ɂ�����Octreotide�̎�ᇐi�W�}�����ʂɂ��ẮA�L���Ƃ���������66)�{�M�ŕی��K���͂Ȃ��B�Ȃ��A������̑g�D�^�ł��̓]�ڂ����݂���ꍇ�́A�ǐ��p�A���W�I�g�Ď܂Ȃǂ̋Ǐ��Ö@���l�����ׂ��ł���56 ,67 ,68)�B
| �\1 P-NET��WHO���� |
 |
| �\2 �X�������ᇂ�WHO�a���g�D����(2000�N�A2010�N) |
 |
| NET�Fneuroendocrine tumor NEC�Fneuroendocrine carcinoma |
| 7���X�X�E���a��(���t���X�E�B��) |
�v��
|
�X�X�E���a�ς̌o�ߊώ@
�Տ��Ǐ�(������̈����Ǐ�Ȃ�)�̂Ȃ��ꍇ�A���Ɍo�ߊώ@�̕K�v�͂Ȃ��B
P-NET�ɑ���o�ߊώ@�ɍۂ��X�X�E���a�ςɂ��Ă��]������B
�X�X�E���a�ς̐f�f����
�Տ��Ǐ�(������̈����Ǐ�)�̏o�����ɐ؏��p���l������B
|
���
VHL�a��7�`71%�̏Ǘ�ɂ������X�X�E���a�ς��݂��A�g�D�^�̔��������Ǘ�ł͂قƂ�ǂ����t���X�E�B��(Serous cystadenoma�FSCA)�ł���69-71)�B�X��SCA�̈������͂����܂�ł���A�X�E�a���傫���Ȃ葼����̈����Ǐ�Ȃǂ̗Տ��Ǐo������܂ŁA�o�ߊώ@���邢�͎��Â̕K�v�͂Ȃ�72)�B�������A���l��VHL�Ǘ�ł́A����������\���̂��鑼���X�X�E���a��(�X�Ǔ������S�t����ᇂ���єS�t���X�E���)�Ƃ̊ӕʂɒ��ӂ��K�v�ł���B
�v��
|
�f�f�͐G�f�ƒ����g�����ɂ���čs���B
��ʂɖ��Ǐ�ł��舫�����̂�����͂Ȃ����Â̕K�v�͂Ȃ��B
|
���
�j�����҂�1�^�A2�^�ɂ�����炸�����p�x��25-60%�̊��҂�10�Α�Ŕ�������B�Б����A�������A�������ł���B����10�~14mm���x�̑傫���ƂȂ�B�Ǐ�Ƃ��Ă͉A�X�̈�a���Ȃǂ�����B�a���I�ɂ͊g���������ǂ̑��ł��葽�����X�E������72)�B�������̏ꍇ�͕s�D�ǂ̉\��������B�������s�D��h�����@�͂Ȃ��B�f�f�͐G�f�ƒ����g�����ɂ���čs��72, 73)�B�ӕʐf�f�͐�����ᇂ���������B�������̉\���͂Ȃ����ߎ�ᇓE�o�Ȃǂ̎��Â͕K�v�Ȃ��ۑ��I�Ȍo�ߊώ@�ł悢�B (��9�� �t���[�`���[�g�Q��)�B
1.�����_�o�n���lj��
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361�F2059-67.
- Ammerman JM, Lonser RR, Dambrosia J, et al. Long-term natural history of hemangioblastomas in patients with von Hippel-Lindau disease: implications for treatment. J Neurosurg. 2006�G105�F248-55.
- Wanebo JE, Lonser RR, Glenn GM, et al. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg. 2003�G98�F82.94.
- Jagannathan J, Lonser RR, et al. Surgical management of cerebellar hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2008�G108(2)�F210-22.
- Lonser RR, Weil RJ, Wanebo JE, et al. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003�G98(1)�F106-16.
- Weil RJ, Lonser RR, DeVroom HL, et al. Surgical management of brainstem hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003�G98(1)�F95-105.
- Ammerman JM, Lonser RR, Dambrosia J, et al. Long-term natural history of hemangioblastomas in patients with von Hippel-Lindau disease: implications for treatment. J Neurosurg. 2006�G 105(2)�F248-55.
- Kanno H, Yamamoto I, Nishikawa R, et al. Spinal cord hemangioblastomas in von Hippel-Lindau disease. Spinal Cord. 2009�G47(6)�F447-52.
- Van Velthoven V, Reinacher PC, Klisch J, et al. Treatment of intramedullary hemangioblastomas, with special attention to von Hippel-Lindau disease. Neurosurgery. 2003�G53�F1306-14.
- Ammerman JM, Lonser RR, Dambrosia J, et al. Long-term natural history of hemangioblastomas in patients with von Hippel-Lindau disease�Fimplications for treatment. J Neurosurg. 2006�G105�F248-55.
- Chang SD, Meisel JA, Hancock SL, et al. Treatment of hemangioblastomas in von Hippel-Lindau disease with linear accelerator-based radiosurgery. Neurosurgery. 1998�G43(1)�F28-34�Gdiscussion 34-5.
- Jawahar A, Kondziolka D, Garces YI, et al. Stereotactic radiosurgery for hemangioblastomas of the brain. Acta Neurochi(r Wien). 2000�G14(2 6�F)641-4; discussion 644-5.
- Kano H, Niranjan A, Mongia S, et al. The role of stereotactic radiosurgery for intracranial hemangioblastomas. Neurosurgery. 2008�G63(3)�F443-50�Gdiscussion 450-1.
- Koh ES, Nichol A, Millar BA, et al. Role of fractionated external beam radiotherapy in hemangioblastoma of the central nervous system. Int J Radiat Oncol Biol Phys. 2007�G69(5)�F1521-6.
- Matsunaga S, Shuto T, Inomori S, et al. Gamma knife radiosurgery for intracranial haemangioblastomas. Acta Neurochir(Wien). 2007�G149(10)�F1007-13�Gdiscussion 1013.
- Moss JM, Choi CY, Adler JR Jr, et al. Stereotactic radiosurgical treatment of cranial and spinal hemangioblastomas. Neurosurgery. 2009�G65(1)�F79-85�Gdiscussion 85.
- Niemela M, Lim YJ, Soderman M, et al. Gamma knife radiosurgery in 11 hemangioblastomas. J Neurosurg. 1996�G85(4)�F591-6.
- Park YS, Chang JH, Chang JW, et al. Gamma knife surgery for multiple hemangioblastomas. J Neurosurg. 2005�G10(2 Suppl)�F97-101.
- Patrice SJ, Sneed PK, Flickinger JC, et al. Radiosurgery for hemangioblastoma: results of a multiinstitutional experience. Int J Radiat Oncol Biol Phys. 1996�G35(3)�F493-9.
- Smalley SR, Schomberg PJ, Earle JD, et al. Radiotherapeutic considerations in the treatment of hemangioblastomas of the central nervous system. Int J Radiat Oncol Biol Phys. 1990�G18�F1165-71.
- Tago M, Terahara A, Shin M, et al. Gamma knife surgery for hemangioblastomas. J Neurosurg. 2005�G102(Suppl)�F 171-4.
- Wang EM, Pan L, Wang BJ, et al. The long-term results of gamma knife radiosurgery for hemangioblastomas of the brain. J Neurosurg. 2005�G10(2 Suppl)�F225-9.
2.���������p�X��
- Choo D, Shotland L, Mastroianni M, et al. Endolymphatic sac tumors in von Hippel-Lindau disease. J Neurosurg. 2004�G100�F480-7.
- Manski TJ, Heffner DK, Glenn GM, et al. Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel-Lindau disease. JAMA. 1997�G277�F1461-6.
- Binderup ML, Bisgaard ML, Harbud V, et al. Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition. Dan Med J. 2013, 60:�@B4763.
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361�F2059-67.
- Kim HJ, Butman JA, Brewer C, et al. Tumors of the endolymphatic sac in patients with von Hippel-Lindau disease: implications for their natural history, diagnosis, and treatment. J Neurosurg. 2005�G102�F503-12.
- Kim HJ, Hagan M, Butman JA, et al. Surgical resection of endolymphatic sac tumors in von Hippel-Lindau disease: findings, results, and indications. Laryngoscope. 2013; 123:�@477-83.
- Lonser RR, Kim HJ, Butman JA, et al. Tumors of the endolymphatic sac in von Hippel-Lindau disease. N Engl J Med. 2004�G350�F2481-6.
- Binderup ML, Gimsing S, Kosteljanetz M, et al. von Hippel-Lindau disease: deafness due to a non-MRI-visible endolymphatic sac tumor despite targeted screening. Int J Audiol. 2013; 52:771-5.
3.�Ԗ����ǎ�
- Dollfus H, Massin P, Taupin P, et al. Retinal hemangioblastoma in von Hippel-Lindau disease: a clinical and molecular study. Invest Ophthalmol Vis Sci. 2002�G43(9)�F3067-74.
- Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999�G117(3)�F371-8.
- Singh AD, Nouri M, Shields CL, et al. Treatment of retinal capillary hemangioma. Ophthalmology. 2002�G109(10)�F 1799-806.
- Aiello LP, George DJ, Cahill MT, et al. Rapid and durable recovery of visual function in a patient with von hippellindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology. 2002�G109(9)�F1745-51.
- Schmidt-Erfurth UM, Kusserow C, Barbazetto IA, et al. Benefits and complications of photodynamic therapy of papillary capillary hemangiomas. Ophthalmology. 2002�G109(7)�F1256-66.
4.���F�זE��
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361(9374)�F2059-67.
- Chen F, Kishida T, Yao M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat. 1995�G5(1)�F66-75.
- Walther MM, Reiter R, Keiser HR, et al. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma.�@J Urol. 1999�G162(3 Pt 1)�F659-64.
- Gimenez-Roqueplo AP, Lehnert H, Mannelli M, et al. European Network for the Study of Adrenal Tumours(ENS@T) Pheochromocytoma Working Group. Phaeochromocytoma, new genes and screening strategies. Clin Endocrinol(Oxf). 2006�G65(6)�F699-705.
- Eisenhofer G, Lenders JW, Linehan WM, et al. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. N Engl J Med. 1999�G340(24)�F1872-9.
- Eisenhofer G, Walther MM, Huynh TT, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001�G86(5)�F1999-2008.
- Eisenhofer G, Huynh TT, Elkahloun A, et al. Differential expression of the regulated catecholamine secretory pathway in different hereditary forms of pheochromocytoma. Am J Physiol Endocrinol Metab. 2008�G295(5)�FE1223-33.
- Maranchie JK, Walther MM. Early identification of patients with von Hippel-Lindau disease at risk for pheochromocytoma. Curr Urol Rep. 2001�G2(1)�F24-30.
- Yip L, Lee JE, Shapiro SE, et al. Surgical management of hereditary pheochromocytoma. J Am Coll Surg. 2004�G 198(4)�F525-34�Gdiscussion 534-5.
5.�t���
- Littrup PJ, Ahmed A, Aoun HD, et al. CT-guided percutaneous cryotherapy of renal masses. J Vasc Interv Radiol. 2007;18(3):383-92.
- Matin SF, Ahrar K, Wood CG, et al. Patterns of intervention for renal lesions in von Hippel-Lindau disease. BJU Int. 2008�G102(8)�F940-5.
6.�X�_�o��������
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361�F2059-67.
- Blansfield JA, Choyke L, Morita SY, et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease(VHL) manifested by pancreatic neuroendocrine neoplasms(PNETs). Surgery. 2007�G142�F 814-8.
- Hough DM, Stephens DH, Johnson CD, et al. Pancreatic lesions in von Hippel-Lindau disease: prevalence, clinical significance, and CT findings. AJR Am J Roentgenol. 1994�G162�F1091-4.
- Yamasaki I, Nishimori I, Ashida S, et al. Clinical characteristics of pancreatic neuroendocrine tumors in Japanese patients with von Hippel-Lindau disease. Pancreas. 2006�G33�F382-5.
- Binkovitz LA, Johnson CD, Stephens DH. Islet cell tumors in von Hippel-Lindau disease: increased prevalence and relationship to the multiple endocrine neoplasias. AJR Am J Roentgenol. 1990�G155�F501-5.
- Hammel PR, Vilgrain V, Terris B, et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d'Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000�G119�F1087-95.
- Langrehr JM, Bahra M, Kristiansen G, et al. Neuroendocrine tumor of the pancreas and bilateral adrenal pheochromocytomas. A rare manifestation of von Hippel-Lindau disease in childhood. J Pediatr Surg. 2007�G42�F1291-4.
- Plockinger U, Rindi G, Arnold R, et al. European Neuroendocrine Tumour Society. Guidelines for the diagnosis and treatment of neuroendocrine gastrointestinal tumours. A consensus statement on behalf of the European Neuroendocrine Tumour Society(ENETS). Neuroendocrinology. 2004�G80�F394-424.
- Reznek RH. CT/MRI of neuroendocrine tumours. Cancer Imaging. 2006�G6�FS163-77.
- Igarashi H, Ito T, Nishimori I, et al. Pancreatic involvement in Japanese patients with von Hippel-Lindau disease: results of a nationwide survey. J Gastroenterol 49:511-16, 2014.
- Triponez F, Goudet P, Dosseh D, et al. French Endocrine Tumor Study Group. Is surgery beneficial for MEN1 patients with smal(1��or=2cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg. 2006�G30�F654-62.
- Lairmore TC, Chen VY, DeBenedetti MK, et al. Duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg. 2000�G231�F909-18.
- Imamura M. Recent standardization of treatment strategy for pancreatic neuroendocrine tumors. World J Gastroenterol. 2010�G16�F4519-4525.
- Hattori K, Teranishi J, Stolle C, et al. Detection of germline deletions using real-time quantitative polymerase chain reaction in Japanese patients with von Hippel-Lindau disease. Cancer Sci. 2006�G97�F400-5.
- Solcia E, Kloppel G, Sobin LH. Histological Typing of Endocrine Tumours, ed 2. WHO International Histological Classification of Tumours. Berlin�FSpringer, 2000.
- Kloppel G, Anlauf M. Epidemiology, tumour biology and histopathological classification of neuroendocrine tumours of the gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2005�G19�F507-17.
- Bosman FT, Carneiro F, Hruban RH, et al. WHO classification of tumor of the digestive system. Lyon�FIARC Press, 2010.
- �X�E�����ǐ_�o�������ᇁiNET�j�f�ÃK�C�h���C���쐬�ψ����. �u�X�E�����ǐ_�o�������ᇁiNET�j�f�ÃK�C�h���C��, ��1��, ���{�_�o�������ᇌ�����, 2013.
- Rinke A, Muller HH, Schade-Brittinger C, et al. PROMID Study Group. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009�G27�F4656-63.
- Sideris L, Dube P, Rinke A. Antitumor effects of somatostatin analogs in neuroendocrine tumors. Oncologist 17:747-55, 2012.
- Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008�G9�F 61-72.
- Libutti SK, Choyke PL, Bartlett DL, et al. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recommendations. Surgery. 1998�G124�F1153-9.
7.�X�X�E���a��(���t���X�E�B��)
- Hough DM, Stephens DH, Johnson CD, et al. Pancreatic lesions in von Hippel-Lindau disease: prevalence, clinical significance, and CT findings. AJR Am J Roentgenol. 1994�G162�F1091-4.
- Hammel PR, Vilgrain V, Terris B, et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d'Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000�G119�F1087-95.
- Neumann HP, Dinkel E, Brambs H, et al. Pancreatic lesions in the von Hippel-Lindau syndrome. Gastroenterology. 1991�G101�F465-71.
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361�F2059-67.
8.������̔X��
- Choyke PL, Glenn GM, Wagner JP, et al. Epididymal cystadenomas in von Hippel-Lindau disease. Urology. 1997�G49(6)�F926-31.
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003�G361�F2059-67.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 9 |
�e��ᇂ̌o�ߊώ@����ю��Ãt���[�`���[�g |
�\ �e�����̌o�ߊώ@�ɂ���(�����J�n����)
���t�@�\��Q������ꍇ�͕���MRI
�@�@�t���A���t�A�X���̉摜�����́A�e�f�ÉȂ̋��͂ɂ��ł������A���Ȃ��ōs���B
�f�f�E���Ãt���[�`���[�g
�f�f�E���Ãt���[�`���[�g
1.�o�ߊώ@�t���[�`���[�g
2.�����t���[�`���[�g
3.���Ó��������@�ڕW�F�@�\��Q���ŏ����ɂ���
�X�N���[�j���O�Ǝ��Ãt���[�`���[�g
�f�f�E���Ãt���[�`���[�g
1.�o�ߊώ@�t���[�`���[�g
2.���Ãt���[�`���[�g
�o�ߊώ@�t���[�`���[�g
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[���O�̃y�[�W��]�@[�����̃y�[�W�̏��]
|
|
|
|
